浙理工团队用O2替代Pt,实现可见光高效催化分解水制氢

▲第一作者:卢楠 ;通讯作者:闫晓庆、刘汶、李仁宏
通讯单位:浙江理工大学、南洋理工大学(新加坡)
论文DOI:https://doi.org/10.1016/j.apcatb.2020.119378
全文速览
可见光催化分解水制氢技术被认为是实现可持续高效清洁生产氢气的重要途径,素有“水中取火”的美誉。本文基于氧气协同的PCET(质子耦合电子转移)效应,利用无Pt助催化剂的g-C3N4材料实现了可见光催化甲醇和甲醛水溶液高效制氢的目标。更为重要的是,我们无需对光催化剂本身进行物理或化学改性,只需简单地调控反应体系的氧分压就能大幅增强光催化产氢速率,从而为高效可见光催化分解水制氢提供了全新的解决方案。
背景介绍
化石能源的持续消耗正不断威胁全球生态系统。改变能源结构,寻找新能源,已成为全人类当前必须面对的首要课题。氢能具有环境友好、无毒无污染、热值高等众多优点,被誉为新时代的“能量货币”。其中,光催化分解水制氢技术是典型的人工模拟光合作用,在过去近半个世纪一直是学术界的研究热点。然而,以甲醇等生物质衍生物作为牺牲试剂的光催化分解水制氢反应不仅包含C-H键活化,还包含了水解离过程,两者反应活化能都很高且需依赖不同的活性位,加之制氢过程会产生少量有毒的CO气体,从而导致目前广泛使用的含Pt助催化剂的制氢体系普遍存在催化剂成本居高不下、催化活性不佳以及稳定性较差的痛点问题。为克服上述光催化制氢领域存在的技术瓶颈,我们有必要开发全新的可见光催化分解水制氢技术。
研究出发点
多相光催化水制氢反应(Photocatalytic Water Splitting, PWS)一般包含三种路径:一是全分解水(Photocatalytic Overall Water Splitting, POWS),即2H2O → 2H2 + O2;二是半分解水(Photocatalytic Partial Water Splitting,PPWS),即 2H2O + sacrificial reagent → 2H2 + oxidation product of sacrificial reagent;三是过渡物种分解水(Photocatalytic Intermediate Water Splitting,PIWS),即2H2O → 2H2 + H2O2。
以上三种方法都涉及了2或4个连续的PCET步骤,都需要跨越相当大的能量势垒,从而导致整个反应进展缓慢。在光催化甲醇等牺牲试剂水溶液制氢过程中,最终生成的H2,其中一个H原子出自甲醇,而另外一个H原子来源于H2O;也就是说,水和甲醇分子都参与了该反应。经典光催化理论认为水分解制氢属于高能垒反应,该类反应的△G > 0(在光催化全分解水生成H2和O2过程中,△G = 237.1 kJ/mol),而且催化反应动力学较慢。但是,光催化氧化分解有机物一般是低能垒反应(△G < 0),催化反应动力学较快,反应过程不可逆;同时,这个过程的决速步骤一般涉及分子氧(O2)的活化以及含氧自由基等过渡物种的生成。因此,我们认识到,如果能基于PCET过程,巧妙地将低能垒催化氧化反应中产生的活性氧物种用于调节高能垒催化制氢过程中缓慢的水解离步骤,进而从根本上改变反应路径,将会是降低生物质小分子水溶液光催化制氢反应能垒、加速光催化动力学过程,最终大幅提高其光催化效率的关键所在。
一般而言,为了提高PWS制氢效率,传统的方法包括改变半导体材料的能带结构(通常通过化学修饰的方法,例如杂原子掺杂和构建异质结结构),以及添加助催化剂以增强质子或者电子的转移,而助催化剂通常是贵金属(如Pt),这就造成了PWS工艺成本过高的难题。为此,许多研究者利用元素掺杂、表面碱化、材料复合和纳米结构设计等技术路径提高光催化活性。然而,改性光催化材料通常存在合成路线复杂、材料结构不可控和催化性能不稳定等诸多问题。因此,我们设想是否存在其他可行的技术路径,在保持光催化剂本征结构的同时,还能进一步增强其可见光催化性能?
图文解析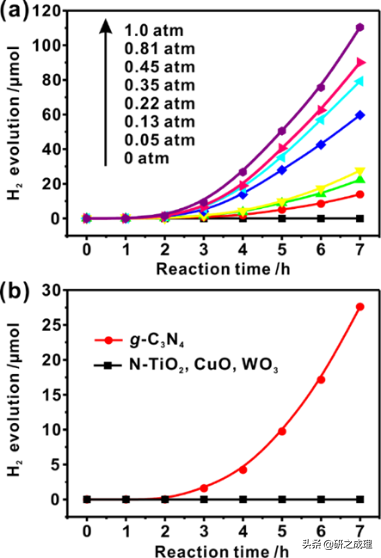
▲图1
在本论文中,我们发现了一种全新、高效以及氧气协同的PPWS反应途径,其基本步骤类似于光催化氧化反应——由分子氧形成活性氧引起,并通过较低能量势垒的基元反应进行。在能带合适的光催化剂上,这种新的PPWS途径可以在有氧条件下由可见光驱动,从而快速制氢。我们发现,分子氧在光催化转化为活性氧之后,作为一种关键的反应介质,可以同时作为氧化剂和均相催化剂参与光催化循环。
传统的光催化分解水制氢系统需要在无氧的条件下进行,而我们的PWS系统的产氢活性却非常依赖O2的含量,并且不需要任何金属掺杂剂或辅助催化剂。初步实验结果表明,在所有的可见光催化实验中,不使用Pt助催化剂的情况下,没有O2或光时并不会产生H2,这表明O2和可见光在促进g-C3N4催化制氢中起着至关重要的作用。
值得注意的是,在甲醇水溶液PPWS中,随着氧分压pO2从0增加到1.0 atm,纯的g-C3N4催化H2析出的速率大大提高,如图1a所示。而在没有O2的情况下,并没有H2从甲醇-水混合物中析出。当pO2 = 1.0 atm时,最佳的H2产率可达到为78.9 μmol h–1 gcatalyst–1。而图1b中可以看到,只有g-C3N4能在氧气的存在下表现出如此可观的光催化制氢活性,其他氧化物基可见光响应光催化剂,如N掺杂TiO2(N-TiO2)、CuO、Fe2O3和WO3完全没有这样的氧气依赖性。
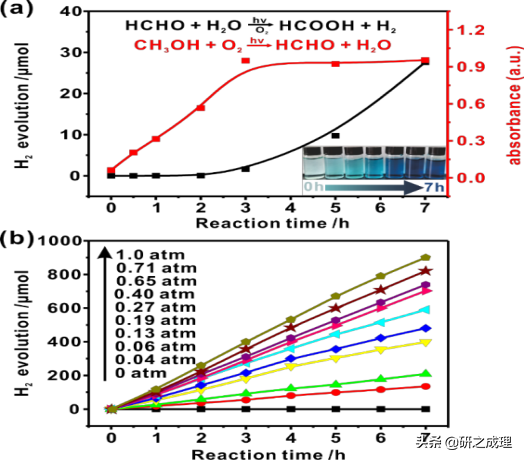
▲图2
我们发现,在氧气的存在下,当光源打开后,并不是立马就有氢气产生的,而是在2小时之后开始产生,即催化制氢反应表现出了明显的“滞后”响应。这种滞后反应表明存在一个或多个限速反应中间体。我们认为甲醇在反应初期被氧气氧化产生了甲醛之类的中间体。因此,我们用酚试剂 法(3-Methyl-2-benzothialinone, MBTH) 和紫外-可见分光度计进行了定性定量的甲醛检测。实验结果如图2a的插图所示,在光反应刚开始的2.0 h内,混合物在MBTH存在下,溶液颜色从近乎透明逐渐变化到深蓝色,则证明了甲醛的存在,并且其浓度不断增加。其紫外-可见分光光谱的测定的最大吸收波长处的吸光度随时间变化的曲线也表明,在反应的前3.0 h,甲醛浓度随时间线性增加,然后达到一个稳定值,而这时氢气也开始以一定的速率开始析出。以上证据表明甲醛确实是甲醇制氢过程中的关键反应中间体。
为了进一步验证甲醛产生的限速作用,我们在其他反应条件相同的情况下,直接将甲醛作为牺牲试剂进行反应。结果表明(图2b),析出的氢气量随时间呈线性增加关系,并不存在任何的诱导期。其催化制氢的速率为643.0 μmol h−1 gcatalyst−1,比甲醇的体系快约8.1倍。因此,甲醇部分氧化为甲醛确实是整个反应的决速步骤,符合在甲醇基PPWS期间观察到的滞后行为(图1和图2)。而与甲醇基体系相类似,甲醛基体系的析氢速率也随pO2的增加而增加,表明分子氧在这两种反应体系中——甲醇氧化和甲醛脱氢,都起着至关重要的作用。
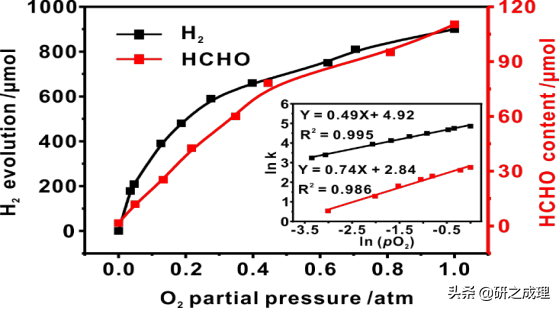
▲图3
当我们作甲醛和H2含量变化与pO2的双对数函数图时(图3),可以得到斜率分别为0.74和0.49的直线,这表明无论在甲醇氧化还是甲醛脱氢反应中,都符合准一级动力学模型。我们还注意到,O2在协同甲醇PPWS过程中是扮演着两个角色的——氧化剂和催化剂。首先,在甲醇光催化制氢过程中,反应器中的pO2随着反应的进行呈逐渐降低态势,即在甲醇部分氧化为甲醛过程中,O2是被作为氧化剂被消耗了的。而在甲醛制氢的过程中,pO2在整个反应过程中几乎保持不变。考虑到促进的H2析出和没有变化的pO2,可以得出结论:O2作为气体催化剂参与了甲醛的重整,而这与我们以前的甲醛非光催化重整工作的结果相似。
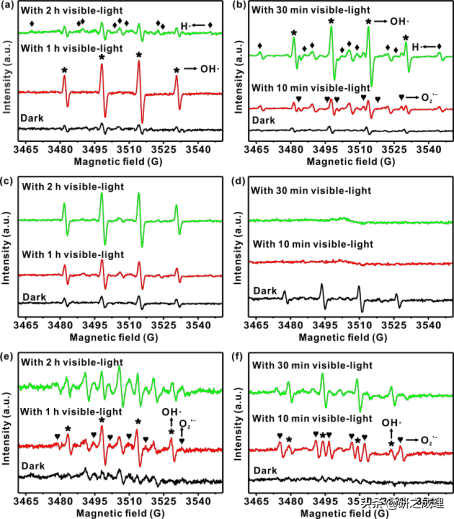
▲图4
我们进一步通过液相EPR自旋捕获法来详细地研究了氧协同的PPWS过程中的反应中间体。在无光照时,含有g-C3N4的碱性甲醇溶液显示了DMPO−•OH加合物的痕迹(图4a)。然而,但测量到的DMPO−•OH加合物是EPR记录过程中经常观察到的副产物,不能被视为真正的反应中间体。之后,在可见光照射一个小时后,随着甲醛浓度达到体系中的临界值,光催化析氢的反应被启动了,水溶液中自由基的种类也发生了相应的变化。DMPO−•H加合物的9重信号峰出现了,这标志着在甲醛析氢过程中氢气是由氢自由基产生的。而甲醛直接用作牺牲试剂时的EPR光谱更验证了这一假设——光照后,•H自由基才开始存在(图4b),而H2能不断析出(图2b)。 此外,我们还检测到一种新的自旋加合物——DMPO−O2•−加合物,表明O2是被还原为超氧物种参与反应的。而在没有O2的情况下,在可见光下,在甲醇反应体系中只检测到DMPO−•OH加合物,而在HCHO体系中观察不到自由基的存在(图4c-d)。此外,在图4e-f中也观察到DMPO−O2•−加合物的存在,但没有检测到•H信号,这就意味着高OH−浓度对底物的活化作用也是必不可少的。根据确定的关键反应中间体和对照实验,我们得出结论,O2首先形成活性氧物种,从而促进水中H2的产生。
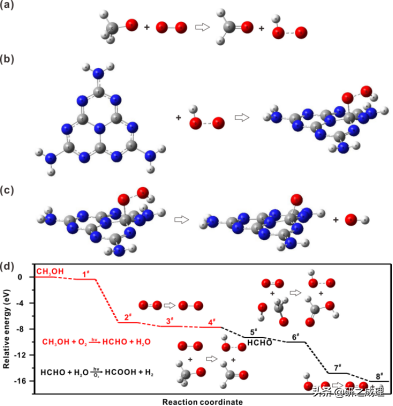
▲图5
根据以上结论,我们辅以DFT计算了所有可能的反应途径(图5),最终得出了一个不同以往的反应机理,共包括了八个基本步骤并仅利用两个电子空穴对:
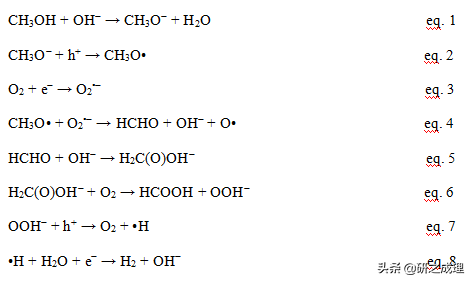
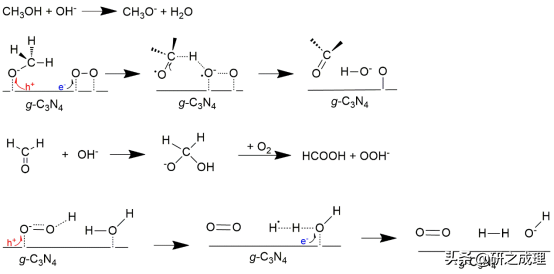
▲图6
首先,OH−将CH3OH分子羟基化为甲氧基阴离子(eq. 1)。由于其亲核性质,产生的甲氧基随后与光生空穴反应生成甲氧基自由基(eq. 2)。同时,O2通过消耗光生电子被还原成超氧离子(eq. 3),随着OH−和O•的产生,甲氧基自由基很容易被还原为HCHO(eq. 4)。而作为一种常见的Cannizzaro反应中间体,HCHO在不需要跨越很大的能量势垒情况下就能羟基化形成H2C(O)OH−(eq. 5)。然后,H2C(O)OH−被氧化为HCOOH的同时形成OOH−阴离子(eq. 6),它可以直接与光生空穴反应形成•H自由基并释放出O2(eq. 7)。最后,在•H自由基的作用下,通过动力学控制对H2O(或质子)进行单电子还原,同时释放OH−阴离子来完成催化循环(eq. 8)。总的来说,八个基本步骤可以分为(I)甲醇部分氧化(eq. 1-eq. 4)和(II)甲醛重整析氢eq. 5-eq. 8),而超氧物种(eq. 3和eq. 4)和氢自由基(eq. 7和eq. 8)分别是两步反应的决速步骤。因此,其能带结构无法将O2还原为O2•−的光催化剂并不适合这种新的氧协同反应途径。图6画出了g-C3N4催化剂、反应物和产物的优化结构,以及整体能量途径。这种新的氧协同的PPWS反应途径完全是放热的,而且几乎没有任何能量势垒。
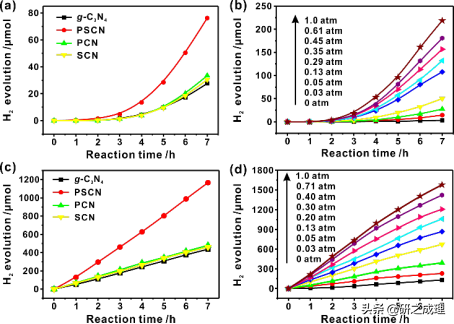
▲图7
之后,我们g-C3N4进行了简单的磷硫共掺杂改性。光催化性能如图7所示,可以看出,所有的掺杂改性后的催化剂在甲醇基体系中均表现出类似于纯g-C3N4的行为:对可见光有着滞后响应,诱导期约2.0 h;随着pO2的增加,制氢速率不断增加。这些相似之处表明,在g-C3N4催化剂上单掺杂P、S或共掺杂P和S之后并没有从根本上改变氧协同的反应途径。而虽然P或S单掺杂能使g-C3N4的PPWS活性分别提高了1.2或1.1倍,但共掺杂P和S的催化剂能使制氢效率提高至三倍。显然,P和S共掺杂是一种成本效益高、易于操作的手段,可以明显提高g-C3N4的氧协同催化PPWS反应的制氢效率。而图7c和7d显示,在光催化甲醛基PPWS时,PSCN在H2产率方面也明显优于其他的光催化剂。而甲醇氧化和甲醛重整都得益于PSCN的光催化活性,这就证实了P和S共掺杂在促进g-C3N4光活性方面的通用性。在pO2=1.0atm时,甲醛基体系在PSCN上的H2产率最大可达1127.3 μmol h−1 gcatalyst−1 (图7c),这种优异的性能远远超过了之前报道的g-C3N4基催化剂,包括那些使用Pt作为助催化剂的反应体系。
▲图8
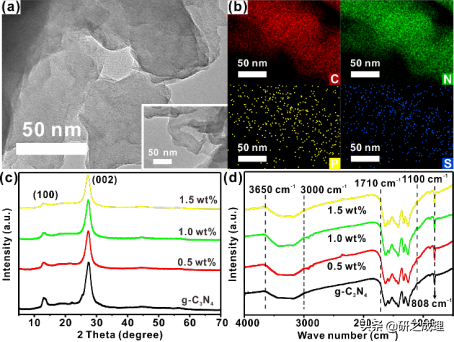
我们对这种掺杂改性能提高催化活性的原因进行了研究。首先,对未掺杂和P、S共掺杂的催化剂形貌结构进行了表征。TEM图像表明,g-C3N4和PSCN均为片状形貌,掺杂之后表面并无明显的纳米团簇存在。而PSCN样品的元素分布能谱显示,P和S元素均匀分布在g-C3N4的二维片层上,这就证明它们已经完全融入到g-C3N4骨架结构中(图8b),而并没有改变其层状形貌。XRD和FI-IR也很好的证明了这点,随着掺杂量的增加,其基本骨架并没有改变。
▲图9
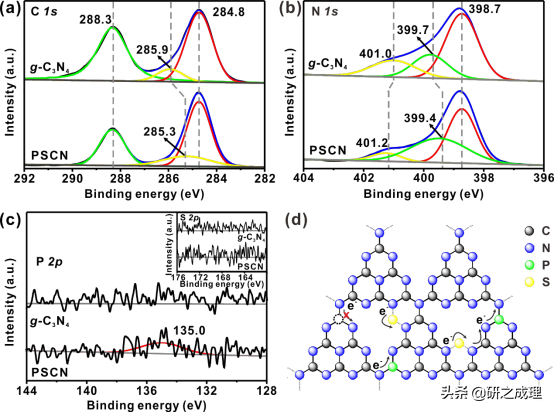
随后,我们用XPS对g-C3N4和PSCN各元素的化学状态进行了检测。在P和S共掺杂后,P2p光谱中在135.0 eV处的存在一个较弱的信号,表明P原子占据了g-C3N4的庚嗪单元中的C位与相邻的N原子形成了P−N键(图8c)。然而,在160−176 eV的结合能范围内没有发现S2p峰(图8c的插图),这表明掺杂的S原子可能通过相对较弱的S−N相互作用占据了平面内七嗪单元之间的间隙位置(图8d)。用电负性较小的P原子取代C原子,能使相邻原子的电子密度变大,从而增强了催化剂结构核层电子的屏蔽效应,使其更适合催化氧还原反应。掺杂后,N1s和C1s峰的位置相对于原始的g-C3N4有了改变,例如,在PSCN中代表N−(C)3键的N1s峰的结合能移到较低的值(399.4 eV)。
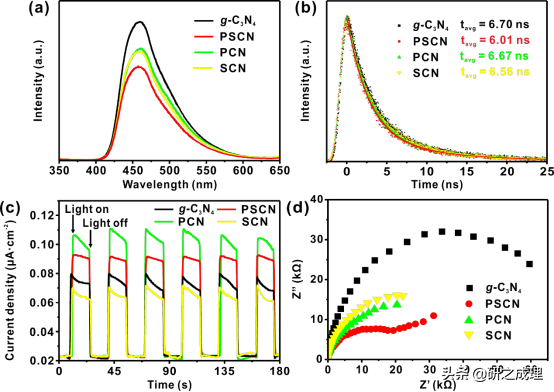
这一变化表明,S和P掺杂能协同调节主体g-C3N4框架的电子结构,并为光生电荷载流子的有效传输提供更多的通道,结构示意图如图8d所示。所以导致了相对于其他g-C3N4基催化剂,PSCN具有最低的光致发光强度(图9a),说明光生电子空穴对的复合在PSCN上减少了,从而增加了量子产率和光催化H2产率。此外,PSCN的时间分辨荧光衰减也表现出最短的寿命,表明光生电子-空穴对的转移在PSCN上比其他三个样品更有效(图9b)。并且光电流响应和电化学阻抗谱也很好的表现了这点。因此,P和S共掺杂会产生协同效应,增强光生载流子的转移,抑制电子空穴对的复合,最终提高量子效率,提高光催化制氢的效率。
▲图10
总结与展望
基于氧气协同的PCET效应,我们发现了全新的可见光催化半分解水反应路径。该反应依赖于体系的氧气分压,达成了单电子分步转移过程,从而实现了低能垒光催化分解甲醇水溶液制氢的目标。当使用g-C3N4这一简单的催化剂时,这一新途径不需任何贵金属作为助催化剂,仅仅使用O2作为反应介质,就能在可见光下高效催化分解水制氢。此外,我们发现在氧化甲醇生成甲醛的过程中,O2充当了氧化剂的角色,而在后续的甲醛重整制氢过程中,O2则作为催化剂参与了该反应。因此,只要增加O2分压,PPWS的效率就可以大大提高。通过P和S共掺杂还可以进一步提高氧气协同的g-C3N4光催化制氢效率,原因在于P和S共掺杂可以在不改变反应途径的情况下促进电荷载流子转移的效率。总之,此类全新的可见光驱动水分解技术不仅证实了在光催化过程中O2具有“氧化剂-催化剂”双功能,而且仅仅使用具有合适能带结构和简单的光催化剂就能开发出高效的光催化水分解制氢系统。
参考文献
[1] X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J.M. Carlsson, K. Domen, M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 8 (2009) 76-80.
[2] R. Li, X. Zhu, X. Yan, H. Kobayashi, S. Yoshida, W. Chen, L. Du, K. Qian, B. Wu, S. Zou, Oxygen-controlled hydrogen evolution reaction: molecular oxygen promotes hydrogen production from formaldehyde solution using Ag/MgO nanocatalyst, ACS Catal., 7 (2017) 1478-1484.
[3] K. Qian, L. Du, X. Zhu, S. Liang, S. Chen, H. Kobayashi, X. Yan, M. Xu, Y. Dai, R. Li, Directional oxygen activation by oxygen-vacancy-rich WO2 nanorods for superb hydrogen evolution via formaldehyde reforming, J. Mater. Chem. A, 7 (2019) 14592-14601.
[4] S. Chen, S. Liang, B. Wu, Z. Lan, Z. Guo, H. Kobayashi, X. Yan, R. Li, Ultrasmall silver clusters stabilized on MgO for robust oxygen-promoted hydrogen production from formaldehyde reforming, ACS Appl. Mater. Interfaces, 11 (2019) 33946-33954.
[5] S. Liang, S. Chen, Z. Guo, Z. Lan, H. Kobayashi, X. Yan, R. Li, In situ generated electron-deficient metallic copper as the catalytically active site for enhanced hydrogen production from alkaline formaldehyde solution, Catal. Sci. Technol., 9 (2019) 5292-5300.
[6] R. Li, X. Zhu, L. Du, K. Qian, B. Wu, S. Kawabata, H. Kobayashi, X. Yan, W. Chen, All-solid-state magnesium oxide supported Group VIII and IB metal catalysts for selective catalytic reforming of aqueous aldehydes into hydrogen, Int. J. Hydrogen Energy, 42 (2017) 10834-10843.
[7] X. Yan, X. Duan, X. Zhou, S. Chen, S. Liang, Z. Guo, L. Du, K. Qian, X. Zhu, R. Li, The interplay of Au nanoparticles and ZnO nanorods for oxygen-promoted, base-free, complete formaldehyde reforming into H2 and CO2, Catal. Commun., 117 (2018) 5-8.
[8] J. Zheng, X. Fu, X. Ying, Y. Zhang, Z. Wang, A sensitive colorimetric high-throughput screening method for lipase synthetic activity assay, Anal. Biochem., 452 (2014) 13-15.
作者介绍
第一作者:卢楠,浙江理工大学化学系硕士研究生,主要研究方向是光催化制氢。
共同通讯1:闫晓庆,博士,浙江理工大学化学系特聘副教授,主要研究方向是光催化制氢和光催化降解有机污染物。
共同通讯2:刘汶,博士,南洋理工大学化学与生物工程教授,主要研究方向是高温反应工程和多相催化,课题组主页:https://www.ntu.edu.sg/home/wenliu/
共同通讯3:李仁宏,博士,浙江理工大学材料科学与工程学院副教授,主要研究方向是催化重整制氢及氢燃料电池。课题组主页:http://mat.zstu.edu.cn/info/1545/3471.html