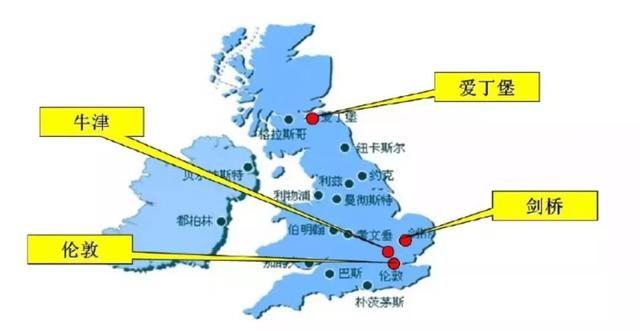
百济取消非小细胞肺癌,与君实、康方抢占鼻咽癌。PD-1海外战升级
一切要比预期的困难。
百济神州替雷利珠单抗合作伙伴诺华发布2022半年财报的同时,也对外披露了更多关于该产品在美国的申报计划。
诺华表示,基于FDA的反馈,诺华及其合作伙伴百济神州已经取消了在美递交非小细胞肺癌(NSCLC)上市申请的计划。不过,下半年两家公司将向美国 FDA 递交替雷利珠单抗一线治疗鼻咽癌的新适应症上市申请。
尽管中国PD-1产品出海此前经历了诸多“考验”,但是如替雷利珠单抗这样明确表示取消一项适应证在美申报的举措还是首次。中国PD-1出海愈发谨慎起来。
01、冲击FDA一波三折
至今,中国创新药企PD-1出海尚无更多振奋人心的消息。
2021年1月,百济神州和诺华宣布达成合作,共同开发、生产和商业化PD-1产品替雷利珠单抗百泽安。据了解,替雷利珠单抗已在35个国家和地区开展了20多项注册性临床试验,入组受试者超过9000人,其中近3000人来自海外。自2021年9至今,替雷利珠单抗先后已经赴美申请了三个适应症上市。
替雷利珠单抗申请的的第一个适应证为用于不可切除或转移性食管鳞状细胞癌(ESCC)患者的二线(2L)治疗。据了解,用于支持这一申请的临床试验为Rationale 302,该试验国际多中心临床,入组512名受试者,其中亚洲人群404人,欧美人群108人。数据显示,替雷利珠单抗治疗组mOS较化疗组显著延长(ITT人群8.6mvs6.3m,PD-L1阳性人群10.3mvs6.8m)且响应比例更高(ORR20.3%vs9.8%)。
但是这一申请的结果尚不明确。根据百济神州今年7月14日的公告内容,针对替雷利珠单抗用于二线ESCC的BLA,FDA因新冠肺炎疫情相关的旅行限制,无法如期在中国完成所需的现场核查工作,因此将延长百该项BLA的审评时间,直至现场核查完成。
百济神州还对外表示,FDA并未对该项BLA的申报材料提出问题。业内人士认为,这通常意味着,企业申报所递交的数据满足审评要求。不过此前,K药和O药均已在美获批了ESCC适应证。
百济神州和诺华这一搭档,针对替雷利珠单抗在FDA申请的第二个上市申请为,用于治疗既往接受过治疗的非小细胞肺癌患者。也是此次宣布取消的适应证申请。
根据诺华首席执行官Vas Narasimhan对外透露的信息,诺华及其合作伙伴百济神州在收到“FDA反馈”后,目前没有申请单药适应证的计划,FDA的结论是,百济神州主导的临床研究“在患者数量和使用的标准治疗方面没有充分反映美国人群特征”,“我认为FDA现在已经非常明确地表明立场,他们希望提交的任何研究都是全球性的,包含了适当数量的美国患者,使用的标准治疗反映了美国的治疗标准。”同时,亦有分析师认为,导致取消申请的主要原因是支持该适应症上市的Rationale-303这一随机、开放性、多中心的全球3期临床试验主要在中美洲和南美,中国和东欧进行。
根据clinicaltrials披露信息显示,该试验覆盖的国家有中国、巴西、保加利亚、立陶宛、墨西哥、新西兰、波兰、俄罗斯、斯洛伐克、土耳其10个国家,共86个临床站点,入组了805例患者,以2:1的比例随机至替雷利珠单抗试验臂或多西他赛试验臂。Rationale-303试验的主要终点为在全部患者(意向治疗患者人群)中以及在PD-L1高表达患者中的OS;关键次要终点包括客观缓解率(ORR)、缓解持续时间(DoR)、无进展生存期(PFS)及安全性。
尽管宣布该适应证取消在FDA的申请,但是今年4月百泽安的上市许可申请(MAA) 获欧洲药品管理局(EMA)正式受理,用于治疗既往接受过全身化疗的晚期或转移性食管鳞状细胞癌(ESCC)患者,以及非小细胞肺癌(NSCLC)患者,这是替雷利珠单抗在欧洲的首项申报。
在非小细胞肺癌适应证的申请上,另一对组合,信达/礼来的信迪利单抗的进展也不顺利。今年3月24日晚间,信达/礼来宣布收到FDA签发的关于信迪利单抗联合培美曲塞和铂类用于非鳞状非小细胞肺癌一线治疗的新药上市申请的完全回复信(CRL)。在完全回复信中,FDA要求信迪利单抗该适应症的申请补充临床试验,建议以总生存期(OS)为终点,进行多地区的临床试验,并与标准疗法头对头对比,采取非劣效的设计。
鼻咽癌是百济神州替雷利珠单抗赴美申请上市的第3个适应证。
鼻咽癌既是亚洲高发适应证,也是美国地区已上市 PD-1 产品未覆盖到的适应证。不过在该适应证在美上市申请上,国内创新药企君实抢先在前。
与百济和信达不同,君实PD-1特瑞普利单抗在赴美上市的适应证申请上,自2021年3月一开始就选择了鼻咽癌这一市场未满足的临床需求。
据了解君实特瑞普利单抗申请FDA上市的依据为 POLARIS-02 的 I/II 期中国区域临床单臂开放式试验及JUPITER-02 的 III 期临床数据(入组新加坡 5 人,中国大陆 270 人,台湾省 14 人)。
招商证券认为尽管该试验方案并非严格的国际多中心(MRCT),但是由于满足了未满足的临床需求,有较大的概率获批。特瑞普利单抗已在黏膜黑色素瘤、鼻咽癌、软组织肉瘤领域获得FDA授予2项突破性疗法认定、1项快速通道认定和3项孤儿药资格认定。
此外,2021年5月,康方/中国生物制药宣布派安普利单抗三线治疗转移性鼻咽癌,已经向美国食品药品监督管理局(FDA)启动提交生物制品许可申请。不过至今尚未有更多进展消息。
在出海适应证的选择上,百济神州高级副总裁、全球药政事务负责人闫小军曾表示,并不会将所有在中国申报的适应证都在美国申报,而是会根据不同适应证的情况进行评估,以支持在美国、欧洲和其他很多国家的申报。
尽管当前中国PD-1产品冲击海外市场尚无定论,但是PD-1国内市场的竞争不容乐观。即将进行的医保谈判中,或将聚齐9个本土玩家参与谈判,同时还有12个新增适应证的谈判。
02、倒逼全球多中心试验水平提升
中国创新药出海注定充满了坎坷,对于企业而言,这是一个漫长的征程,需要企业愈挫愈勇。
中国PD-1在FDA的一系列经历,也在倒逼中国药企提升国际多中心临床试验的掌控能力和组织能力。业内人士认为,如果一款药能够在与FDA的沟通中完成三期临床试验,甚至走得更远,那对于中国药企的收获是无价的。
尽管影响药品国际化注册效率的因素颇多,但是关键还是看产品,如果是差异化的产品或者是first-in-class或者best-in-class,在国际化注册时可发挥的空间相对较大。
招商证券建议,对于药企而言,若以“unmet medical need”策略出海申请 FDA 批准上市,不仅可以大幅缩短临床时间,依靠 I/II 期临床数据上市,同时会大幅节约需要投入的研发开支。数据显示,2010~2022年FDA批准的440 种创新药上市的临床实验时间的中位数为 8.3 年。而百济神州的泽布替尼复发难治的套细胞淋巴瘤 MCL 适应证、传奇生物的西达基奥仑赛 5 线多发性骨髓瘤适应症获得FDA批准上市,分别仅有了4年多时间。
泽布替尼是首个美国FDA主要基于来自中国的数据批准的来自中国制药企业的产品、首个在中美均获得加速批准上市的产品、首个先在美国获批后在中国获批的国产产品。
2015 年 3 月,百济神州泽布替尼用于2线治疗复发难治的套细胞淋巴瘤(MCL),开始与FDA 进行pre-IND沟通,2019 年6月递交DNA,2019 年 8 月获得优先评审资格,当年11月获得 FDA批准上市,仅用了4年半时间。
据了解,对于该产品,百济神州自立项之初就已经有出海的计划和打算。在临床申报时,中国和澳洲同步进行,并在之后不久,就开启了与FDA的沟通,随后在美国开启临床试验。
闫小军介绍,在与FDA首次面对面的Pre-IND meeting之前,百济神州做了充分的准备,“真的是一个单词一个单词、一句话一句话地去推敲我们所有的问题,和我们自己对于每个问题的观点、怎么回答等等,团队做了很多的演练”。
而传奇生物的西达西达基奥仑赛改变了多发性骨髓瘤后线治疗无药可用的境况。2018 年 5 月西达基奥仑赛获得IND许可,2019年12月获得突破性疗法认证,2020年12月递交 BLA,2021年5月被授予优先评审资格,2022年2月获批上市,总耗时不到 4 年。
西达基奥仑赛5线多发性骨髓瘤适应症获批依据为 CARTITUDE-1 临床,CARTITUDE-1为 Ib/II 期单臂临床试验,计划入组美国及日本患者128人,II 期临床试验主要临床终点为ORR,12.4 个月随访期,总缓解率ORR高达 97%,sCR高达67%,随访2年治疗获益进一步增加,ORR高达97.9%,sCR高达82.5%。
但是对于非“unmet medical need”项目,招商证券表示,与前期出海路径探索不同,国内创新药企业在最近申请的III期临床项目过程中,已经意识到严格遵守临床试验标准的重要性,并且逐渐明晰海外审批规则并予以遵照。
例如,百济神州PD-1+TIGIT 针对1线 NSCLC适应证的AdvanTIG-302 临床试验中,根据clinicaltrials数据,该试验计划入组605人,224个临床中心,其中美国17个,除中国及中国台湾地区之外,试验还覆盖了15个国家和地区。对照组别设置为目前标准疗法 K药,同时有TIGTI单药组别。
本文相关内容参考自:
1.招商证券《FDA 审批新药标准下的中国创新药企临床策略与标的筛选》报告
2.Endpointsnews,Nicole DeFeudis,Novartis scraps key PD-1 submission, while touting 'significant bolt-on M&A firepower'