陈俊丰团队Nature:不对称缩合构建硫立体中心
本文来自微信公众号:X-MOLNews
多样性导向合成(diversity-oriented synthesis, DOS)可有效地构建结构复杂且功能多样的化合物库,在药物研发中有着广泛的应用。立体异构化合物具有不同的构型特征,常常与靶蛋白产生不同的相互作用。目前在药物研发中占主导地位的无疑是基于碳立体中心的多样化分子骨架,它们为药物研发提供了广阔的化学空间,而相比之下,硫立体中心作为药效团却经常被人们所忽视。近年来,含有S(VI)立体中心的亚砜亚胺(Sulfoximine)凭借独特的理化性质和药代动力学性质,已成为药物研发领域中的一颗新星。尽管还没有含亚砜亚胺的候选药物被批准,但AZD6738和BAY 1000394等几种化合物已进入临床试验(图1a)。受亚砜亚胺所取得进展的启发,不少科学家对其它硫立体中心产生了广泛的兴趣,如亚磺酸酯(sulfinate ester)、亚磺酰胺(sulfinamide)、磺酰亚胺酯(sulfonimidate ester)、磺酰亚胺酰胺(sulfonimidamide)等(图1b)。尽管研究者已经开发了一些新方法来实现硫立体中心的外消旋合成,但要想对映选择性合成硫立体中心仍然是一项艰巨的挑战。手性亚磺酸酯可由廉价易得的手性醇衍生而来并且易转化为其它硫立体中心,因此在硫立体中心化合物中占据关键的位置。然而,有关手性亚磺酸酯的构建报道很少,基本都基于肽或金鸡纳生物碱催化的亚磺酰氯与醇的动态动力学拆分(图1c)。因此,开发一种适用于不同硫立体中心药物后期修饰的催化方法,就显得非常迫切。
近日,新加坡南洋理工大学的Choon-Hong Tan(陈俊丰)教授(点击查看介绍)团队利用盘扭五氮胍盐(Pentanidium,PN)作为手性催化剂实现了亚磺酸钾与醇的不对称缩合,构建了一系列不同的手性亚磺酸酯,为制备亚磺酸酯及其相关的硫立体中心提供了一种可行且统一的合成策略(图1d)。该反应不仅条件温和、官能团耐受性好,而且还能实现塞来昔布、其它上市药物以及药物中间体的后期多样化修饰,并通过硫的立体中心取代磷的立体中心来补充ProTide策略。Xin Zhang博士和Choon-Hong Tan教授是本文的共同通讯作者。相关成果发表在Nature 上。

图1. 手性硫化合物的合成及其药效价值。图片来源:Nature
盘扭五氮胍盐(PN),是陈俊丰教授团队在2011年报道的一种sp2杂化氮中心、胍为骨架的不对称相转移催化剂(J. Am. Chem. Soc., 2011, 133, 2828)。使用这种催化剂,陈俊丰教授团队完成了一系列漂亮的工作(点击阅读详细),包括发表在Science 上的不对称亲卤亲核取代的SN2X反应(Science, 2019, 363, 400, 点击阅读详细)。
回到这项工作,作者首先选择4-甲基苯亚磺酸钾1为模板底物对反应条件进行筛选(图2)。结果显示不同类型的酰氯(2a-2g)和磺酰氯(2h-2i)均可生成中间体混合酸酐,随后硫立体中心被乙醇取代得到所需的亚磺酸酯4(Entry 1-9),其中氯甲酸乙酯2a的效果最好。而对盘扭五氮胍盐(PN)进行的筛选表明:Ar2上含有一个酚羟基的PN1具有较好的立体选择性,若羟基被保护(PN3和PN4),对映体选择性则显著降低(Entry 11-12),这主要是由于PN1的酚羟基与亚磺酸盐1之间的选择性氢键作用。此外,向反应中加入硫醇盐(3a-3d)等添加剂也能促进该反应的发生,特别是3c能以优异的收率(96%)和对映选择性(96%)得到目标产物4,同时还能以克级规模进行制备。
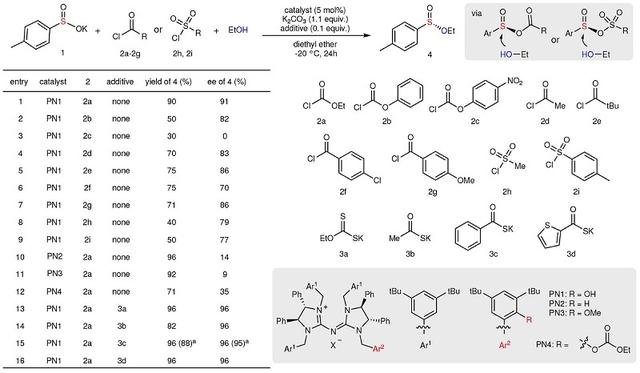
图2. 反应条件的筛选。图片来源:Nature
在最优条件下,作者对苯亚磺酸钾1的底物范围进行了探索(图3)。结果显示不同取代模式的富电子(如:烷氧基(5-7)、烷基(8-9)、均三甲苯基(10)、对乙酰氨基(11))及卤素原子(13-18)取代的苯亚磺酸钾均能兼容该反应,以良好的收率和优异的立体选择性获得所需的手性亚磺酸酯;但吸电子基团取代的苯亚磺酸钾(如:2-三氟甲氧基(20)、氰基(21))的对映选择性却有所降低。此外,不同基团取代的亚磺酸萘酯(22-24)、噻吩和苯并噻吩(25-29)亚磺酸酯甚至烷基亚磺酸盐(30-33)均能以优异的立体选择性实现这一转化。最值得注意的是,PN1与富电子底物反应时会快速形成PN4,导致产率和立体选择性降低,这可通过向反应中加入碱K2HPO4并增加催化剂或添加剂的量来解决这一问题。
其次,该方法对于具有官能团多样性的醇同样适用,例如:(S)-缩水甘油以98:2 的非对映异构体比获得亚磺酸酯(34),并且不影响环氧基团;(R)-1,3-丁二醇进行反应时,伯醇优于仲醇,从而以97:3的dr值得到单亚磺酰化产物(35)。需要指出的是,该方法对于核苷的修饰同样适用,并以中等至高产率和优异的立体选择性获得所需的核苷亚磺酸酯(36-42)。有趣的是,该反应还能实现抗病毒药物齐多夫定、索非布韦和瑞德西韦等中间体以及几种生物活性环状醇(如:胆钙化醇、胆固醇、表雄甾酮和薄荷醇)的立体选择性亚磺酰化,并以中等的收率和较好的立体选择性获得相应的手性亚磺酸酯(39-48)。对胆固醇和薄荷醇的研究表明,若使用ent-PN1 为催化剂,则非对映异构体的比例会反转,表明该反应是催化剂控制而不是底物控制。此外,该方法还适用于伯醇和仲醇,包括异丙醇;而体积庞大的叔丁醇、酚类和胺类并不反应。
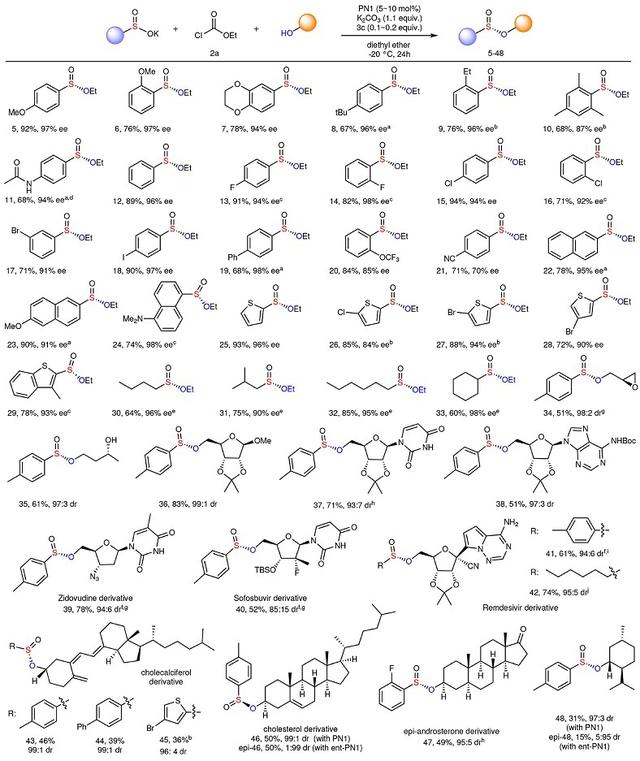
图3. 底物拓展。图片来源:Nature
为了证明该方法的通用性和有效性,作者从药物或药物中间体出发制备了几种复杂的亚磺酸盐,并在不对称缩合条件下构建手性亚磺酸酯。例如:1)西地那非经磺酰化、制备亚磺酸盐(49)、不对称缩合三步便能以高对映选择性得到亚磺酸西地那非酯(50,图4a);2)Etoricoxib的甲基砜经烷基化和苯乙烯的原位消除(51)、不对称缩合获得Etoricoxib亚磺酸酯(52,图4b);3)生物活性伯磺酰胺经卡宾催化脱氨转化为亚硫酸盐,后者经不对称缩合获得相应的亚磺酸酯(53-55,图4c),如:(S)-舒必利、格列本脲和Valdecoxib亚磺酸酯。
鉴于亚磺酸酯是药物后期多样化修饰形成大量硫立体中心的理想关键中间体,作者从塞来昔布出发(图4d、4e),先将其转化为塞来昔布亚磺酸盐(56),随后分别与胆固醇或2-丙炔-1-醇进行不对称缩合,得到所需的塞来昔布-胆固醇亚磺酸酯(57)和炔丙基亚磺酸酯(59)。需要指出的是,59可与齐多夫定上的叠氮基发生“点击反应”获得塞来昔布-齐多夫定络合物(60)。另外,高对映选择性的塞来昔布亚磺酸酯(58)作为S(IV)/S(VI)立体中心的通用前驱体,其硫立体中心能够被各种亲核试剂取代。例如:1)甲基格氏试剂和烯酸锂分别与58反应获得手性纯亚砜(61、62);2)双(三甲基硅基)氨基锂(LiHMDS)进行反应时则得到未保护的亚磺酰胺(63);3)伯胺或仲胺通过形成氨基锂或被格氏试剂活化均可与(58)反应获得高对映选择性的亚磺酰胺(64-66)。最后,作者对塞来昔布亚磺酸酯(58)、塞来昔布亚砜(61)和塞来昔布亚磺酰胺(66)进一步亚胺化,以高收率和高对映选择性得到相应的产物(67-69),而先前的方法则难以实现S(IV)/S(VI)立体中心转化。
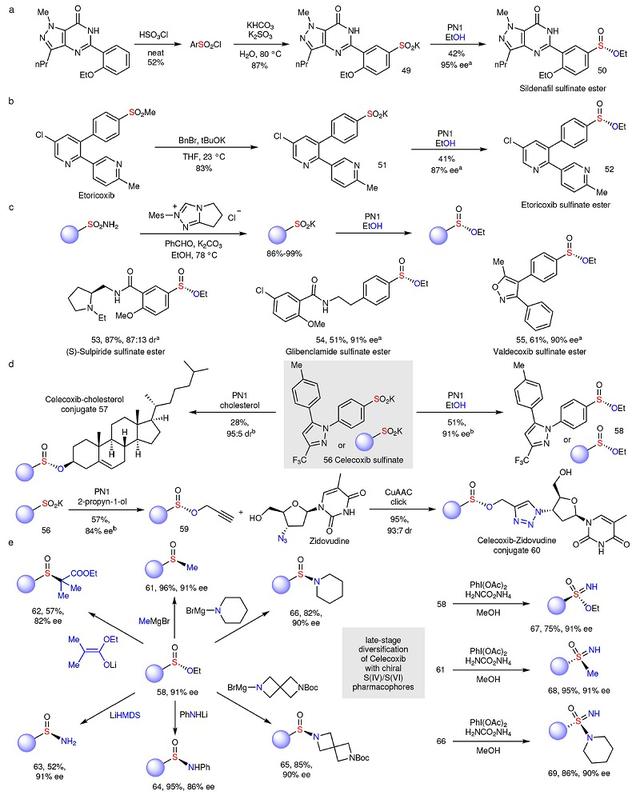
图4. 药物分子的后期修饰。图片来源:Nature
总结
Choon-Hong Tan教授和Xin Zhang博士等研究者利用盘扭五氮胍盐催化剂实现了亚磺酸钾的不对称缩合以构建手性亚磺酸酯,并为制备亚磺酸酯及其相关的硫立体中心提供一种可行且统一的合成策略。该方法不仅条件温和、官能团耐受性好,而且还能实现塞来昔布、其他上市药物以及药物中间体的后期多样化修饰,并通过硫的立体中心取代磷的立体中心来补充ProTide策略。鉴于硫立体中心作为药效团的使用前景看好,相信这种新方法将为药物研发工作者提供更多选择,有助于进一步探索含硫药效团的作用。
Synthesis of chiral sulfinate esters by asymmetric condensation
Xin Zhang, Esther Cai Xia Ang, Ziqi Yang, Choon Wee Kee, Choon-Hong Tan
Nature, 2022, DOI: 10.1038/s41586-022-04524-4
导师介绍
陈俊丰
https://www.x-mol.com/university/faculty/4457
(本文由吡哆醛供稿)




















评论