继Science, Nature子刊和Joule之后,电催化合成H2O2再发Chem

DOI:10.1016/j.chempr.2019.12.008
本文亮点
酸性介质中的电化学氧还原反应(两电子ORR)为直接产生过氧化氢(H2O2)和现场应用提供了一种有吸引力的途径。然而目前仍然缺乏具有高催化性能且低成本的电催化剂。近日新加坡南洋理工大学刘彬课题组与大连化物所黄延强研究员团队合作,从理论上设计和实验证明,锚定在氮掺杂石墨烯中的钴单原子催化剂(Co-SAC)具有最优的*OOH中间体吸附能,表现出很高的H2O2生成率,甚至略优于最先进的贵金属基电催化剂。在0.1M HClO4中0.6V(相对于可逆氢电极)下Co-SAC上生成H2O2法拉第效率 > 90%,动力学电流可达1mA / cm 2。进一步的动力学分析和原位同步X射线吸收研究与DFT计算相结合表明,氮配位的单原子Co是活性位点,并且反应速率受到第一步电子转移步骤的限制。
背景介绍
过氧化氢(俗称双氧水,H2O2)作为一种环境友好的氧化剂,其应用范围广泛。当前的H2O2工业合成方法涉及高能耗的蒽醌氧化/还原步骤,需要复杂的大型设备,同时会产生大量废物。而在燃料电池装置中通过电化学反应过程直接生产H2O2(阳极:H2→2e-+ 2H+;阴极:O2+ 2e- + 2H +→H2O2,E0 =0.695 V),提供了一种有前景的替代途径。其中阴极的氧还原反应(ORR)通过两电子途径发生。近年来,在碱性介质中通过两电子ORR来产H2O2取得显著进展。最近的结果表明,进一步改善碱性条件下催化剂(主要为碳基材料)的活性和选择性的空间很小。但是,在碱性介质中生产H2O2有几个缺点:(1)H2O2的稳定性较差,可以在碱中自发分解(尤其是在pH >9时);(2)碱性条件下的阴离子交换膜,其稳定性和电导率远不及用于酸性条件的质子交换膜;(3)H2O2在酸性介质中具有比碱性介质更强的氧化能力。例如,芬顿试剂主要用于有机合成和废水处理,其最适pH范围为2.5 – 3.5。因此,研究酸性介质中的H2O2催化合成更有意义。先前的研究(Nat. Mater. 12, 1137-43)表明碳载汞铂合金或汞钯纳米颗粒用于酸性介质中通过两电子ORR合成H2O2,表现出高活性和选择性。但是,这些催化剂包含稀有的贵金属和有毒的汞,因此限制了它们在H2O2生产中的潜在应用。尽管均相分子催化剂(例如钴酞菁)对于通过ORR生产H2O2具有高度选择性,但其低活性和低稳定性导致难以应用。总之,仍然缺乏在酸性介质中通过两电子氧还原反应合成H2O2的高性能低成本催化剂。
近年来,具有明确定义活性中心的单原子催化剂(Single AtomCatalyst)在多种化学反应中表现出高活性和高选择性,引起了科研人员的极大关注。从原理上来说,通过ORR来生产H2O2,首先必须尽可能避免O-O键在催化剂表面断裂。得益于单原子催化剂的特征,其中活性位点是单个的,相互隔离的,因此O2在单原子催化剂上的吸附通常是end-on 类型,而不是μ-peroxo 配位,因此可以减少O-O键断裂的可能性。也就是说单原子催化剂很可能适合两电子ORR生成H2O2。
通过DFT理论计算和实验方法相结合,本文系统地研究了过渡金属单原子催化剂(Mn,Fe,Co,Ni和Cu)的结构与两电子ORR合成H2O2的催化性能之间的关系。理论上预测的活性火山图的与实验结果均表明,Co单原子催化剂具有最佳的d带中心,合适的*OOH中间体吸附能,可作为酸性介质中合成H2O2的高活性和高选择性催化剂,其性能甚至略优于最先进的贵金属基催化剂。
结果与讨论
图1. DFT 理论计算.
首先通过DFT计算研究了锚固在氮掺杂石墨烯中的各种过渡金属M-SAC(M=Mn,Fe, Co, Ni, Cu)分别沿2e-或4e-途径产生H2O2或H2O的ORR工艺。经由ORR生成H2O2的双电子(2e-)路径包括两个质子偶联的电子转移步骤,其中只有一个中间体(*OOH)。而对于4 e-ORR途径,包括四个质子耦合电子转移步骤,其中O2依次还原为*OOH,*O,*OH和H2O,如图1A所示。从理论上讲,用于合成H2O2的理想催化剂应使步骤(1)和(2)的动力学阻碍最小化,以提供高活性。同时,催化剂需要尽可能避免*OOH的解离或进一步还原为*O和*OH,以实现高选择性。图1B显示,*OOH,*O和*OH的结合能与M原子中的价电子数几乎成线性下降关系。金属M中的价电子数量越多,这些中间体与M原子的结合越弱,这是因为M原子的d带中心相对于费米能级从Mn到Cu逐渐降低(图1B)。详细地讲, M原子的d轨道与键合的O原子的2p轨道之间的偶合所产生的反键轨道能量从Mn到Cu逐渐下降。反键轨道被填充得越多,金属M-O键越弱。为了比较这些SAC的ORR活性,我们计算了自由能图,并使用ΔG*OH作为描述符构造了2e和4eORR路径的活性火山图,如图1C 所示。对于理想的2e- ORR催化剂,*OOH的吸附热应在平衡电势下(U = 0.7 V vs RHE)是零,对应于ΔG*OOH=〜3.5±0.2 eV. 然而与2e-ORR不同的,即使对于最优的4e- ORR催化剂,由于*OOH和*OH吸附能之间的比例关系(scalingrelationship, 即ΔG* OOH = 0.747ΔG* OH + 3.32 eV),也需要〜0.4 V的超电势才能将O2还原为H2O.这在其他很多模型中也发现了类似的结果。从图1C可以看出,Ni和Cu-SAC上的ORR更倾向于走2e-途径到H2O2,但由于较大*的OOH还原势垒和较高的O2活化能垒(图1D),这两种催化剂将显示出较低的ORR活性但对H2O2的产生应该具有较高的选择性。
相比之下,O2在Mn和Fe-SAC上的吸附很强(图1D),以至于在将*OOH还原为*O时,热力学上变得容易。因此,ORR在Mn-和Fe-SAC上将以 4e-途径为主,因此对于H2O2的选择性会很低。此外,由于Fe-SAC具有4e- ORR最优吸附能(图1B和C),Fe-SAC应该在这五种单原子催化剂中具有最高的4e- ORR活性。而具有最佳d带中心的Co-SAC在U = 0.7 V时ΔG* OOH = 3.54 eV,对OOH的吸附既不太强也不不太弱,几乎位于2e- ORR火山图顶点(图1C)。因此推测Co-SAC对于2e-生成H2O2具有很高的活性。此外,与Mn-和Fe-SAC相比,Co SAC上将*OOH还原为*O中间体的势垒更高,将提高Co-SAC对H2O2的选择性。结合预测的高活性,可以推测,Co-SAC上H2O2的产率将是五个单原子催化剂中最高的。
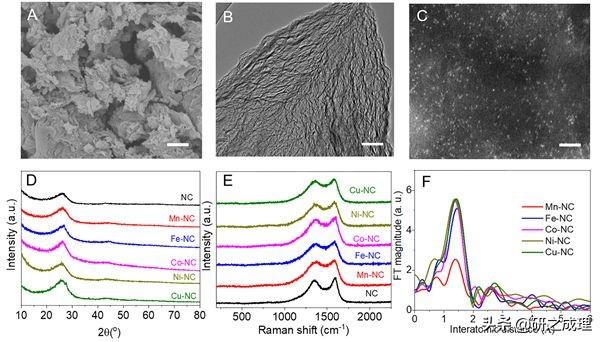
图2.五种单原子催化剂的表征(M-NC,M=Mn, Fe, Co, Ni, Cu)
随后,我们通过三聚氰胺,L-丙氨酸和相应的金属盐混合物的热解成功合成了五种不同的过渡金属原子催化剂。图2及其他表征手段证明合成的单原子催化剂结构与计算中用的模型结构基本一致。

图3.五种单原子催化剂生成H2O2的性能测试.
接下来,在旋转环盘电极(RRDE)上于0.1 M HClO4中进行了五种单原子催化剂生成H2O2的性能测试。图3A显示Co-NC和Fe-NC对ORR的活性更高,其起始电位约为0.7V。Co-NC的环电流(对应于H2O2的氧化)也在0.7V左右开始,计算出的H2O2法拉第效率(图3B)表明,Co-NC在整个电位范围内对H2O2的生产具有高度选择性。在0.6V电压下,Co-NC上生成H2O2的动力学电流达到1mA / cm2,法拉第效率> 90%。而Fe-NC对H2O2的选择性低得多,与上面DFT预测的一致。尽管其他催化剂Mn-NC,Ni-NC,Cu-NC和NC由于对H2O2具有很高的选择性(图3B),它们的催化活性很差。以上实验结果与我们的DFT计算结果基本一致(图1),即Co-NC位于H2O2生成反应的活性火山图顶部。图3C比较了通过ORR生产H2O2的电催化剂的性能。可以看出,Co-NC是H2O2合成最有效的催化剂,甚至略胜于以前在酸性介质中报道的最好的Pd-Hg合金催化剂。除活性外,稳定性是催化剂在实际使用中的另一个重要考虑因素。在静态和旋转条件下,研究了Co-NC在0.1 V HClO4中0.5 V下的稳定性。环形电极和圆盘电极的电流在10小时内保持稳定,没有明显的衰减(图3D);环形电极的电流略有增加可归因于电解液中逐渐积累的H2O2。在整个过程中,通过滴定法确定的H2O2选择性仍然高达〜88%。

图4.反应过程机理分析.
从DFT计算中,我们发现第一步(* + O2 + H + + e-→* OOH)是Co-NC上ORR的热力学过电势决定步骤。为了更深入地了解反应机理,我们进行了动力学分析,以实验方式确定速控步。首先,通过Koutecky-Levich分析,从在不同转速下获得的LSV曲线获得了Co-NC上ORR的动电流。通过在不同的O2分压下进行ORR来确定O2的反应级数。图4A显示了在不同的O2分压下动电流与超电势的关系。然后,如图4B所示,绘出了动力学电流的对数与O2分压的对数关系,由此可以推导出O2的反应级数(线的斜率)在过电势从0增加到250mV时从0.53增加为0.90。同时,随着过电势从0 mV升高到250 mV,Tafel斜率从〜110 mV dec-1增加到〜140 mV dec-1,最后达到〜240 mV dec-1(图4A)。图4C和4D显示了pH(H+离子浓度)对Co-NC上产生H2O2活性的影响。通过与图4A和4B相同的分析,得出H+的反应级数为 -0.05〜-0.07,非常接近于零,如图4D所示。说明在决速步骤中不涉及H+,即质子化过程应进行得很快。通过组合图4A-D和表1,可以得出在Co-NC上合成H2O2的速率决定步骤为:*+ O2 + e-→* O2-,该步包含在DFT预测的热力学过电势决定步骤中。
表1.可能的决速步及对应的理论Tafel斜率,O2,H+的反应级数

详细来说,在相对低的过电势(<50 mV)下,它主要由吸附O2的电子转移步骤控制。(*O2 + e-→*O2-)。通过增加过电势,电子转移步骤变得更快。然后,总反应速率更多受到O2吸附过程的限制,这与观察到的逐渐增加的Tafel斜率和O2的反应级数吻合。另外,为了监测Co电子态和配位环境的变化,进行了原位同步X射线吸收光谱研究,空气中Co-NC的EXAFS光谱与浸入充满N2的电解质中的Co-NC的光谱几乎相同,两种情况下Co-N的距离均为1.25Å。将N2切换为O2后,观察到傅立叶变换(FT)强度显着增加,Co-N距离略微增加至1.35Å,表明O2在Co原子上的吸附将Co原子轻微拉出平面。该结果与DFT计算的氧物种应吸附在Co原子的顶部结论一致。在0.6V,部分吸附的O2通过ORR转化为H2O2,Co-N距离减小至1.32Å(图4E)。在较低的电势下(0.3V),由于O2的吸附速率有限,而吸附的O2通过ORR转变为H2O2的速度较快,此时O2的表面覆盖率变得更低,从而Co-N距离进一步减小到1.29Å。在电势返回到开路电势下,Co-N键长恢复到其初始值(1.35Å),并且k空间中的EXAFS光谱也显示出类似的趋势。这种趋势与动力学分析非常吻合。图4F显示了在Co-NC上进行H2O2生产的ORR步骤的示意图,其中步骤2是在较高电势下的速率限制过程,而步骤1在较低电势下成为速率限制过程。形成*OOH后,迅速进一步还原为H2O2,并且通过H2O2的解吸使活性位点再生,从而完成了催化循环。