深度长文:生物制剂管线全解析
深度长文:生物制剂管线全解析!
来源:药智网|辛铃

当所有人都认为COVID-19大流行可能会严重影响2020年的药物批准时,行业和监管机构却创造了一个小小的奇迹。截至12月31日,美国食品和药物管理局(FDA)在2020年批准的药物和生物制剂数量几乎与2019年一样多(图1和表1)。
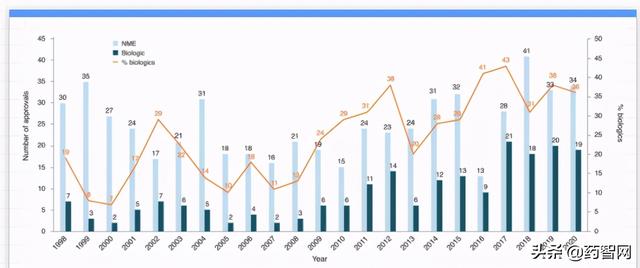
图1 2020年美国FDA批准的药物数量
(NME:新分子实体,biologics:生物制剂)
尽管2020年COVID-19大流行,但药品监管机构似乎没有错过任何一款创新药的审批机会。针对罕见疾病和遗传性癌症治疗,2020年美国FDA批准了多款新药,包括增加了一种嵌合抗原受体(CAR)T细胞疗法Tecartus,药物设计采用了更“纯净”的制造工艺;同时,在重磅级别的慢病适应症上,全球首次批准了一款小干扰RNA(siRNA)新药——欧洲药物管理局(EMA)批准了RNA干扰(RNAi)疗法Leqvio(inclisiran)用于治疗心血管疾病及高胆固醇症。
表1 2020年美国FDA批准的生物制剂
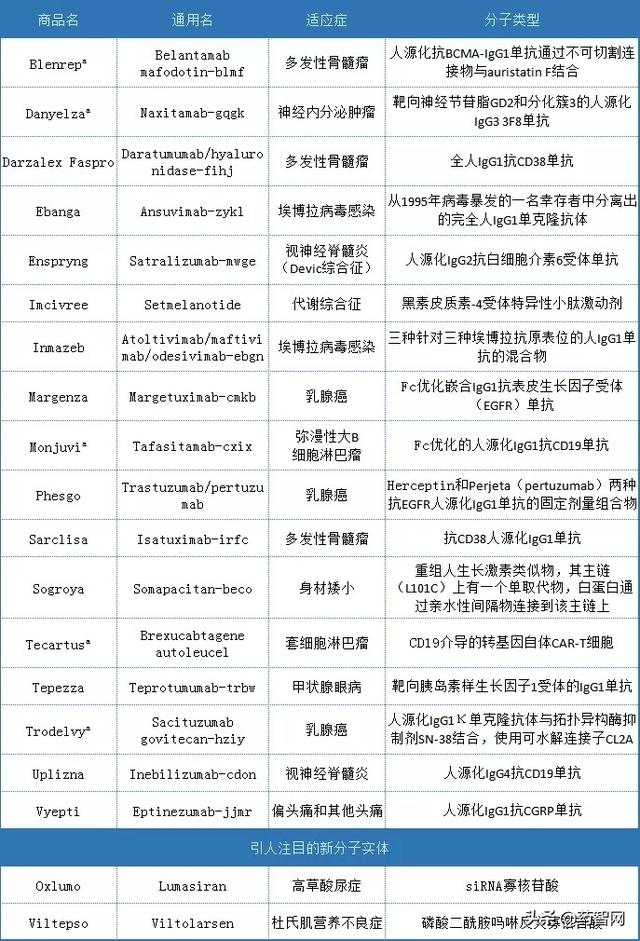
(注:a 代表加速审批)
除此之外,2020年监管审批的另一亮点是针对COVID-19药物的快速批准和紧急使用授权(EUA)。美国FDA在COVID-19大流行期间发布了多项EUAs,主要用于体外诊断和个人防护设备,但也有一些用于治疗和预防(表2)。EUA不是批准,它只在紧急情况期间有效,审查也不那么严格。尽管美国FDA的常规批准会提及安全性和有效性的“实质性证据”,但EUAs是基于合理的信念发布的,即产品可能是安全和有效的。
表2 美国FDA对抗COVID-19发布的紧急使用授权(EUA)
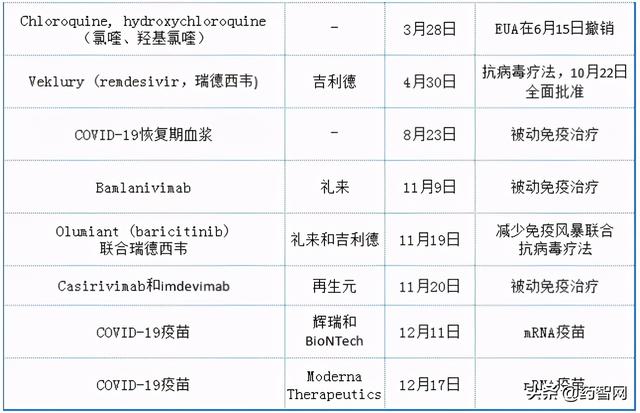
来源:美国FDA
通过一年的实践,EUA给监管机构和行业也留下了宝贵的经验教训,可能成为药物开发未来的指导:
一、监管过程根据普遍的压力而变化(这一教训实际上适用于EUA和完全批准)。事实证明,与监管机构的面对面会议不如定期交换信息的机会重要。对于COVID-19相关的产品,监管机构更重视速度的需要,考虑了并行而不是顺序的监管步骤。
二、并不是每个EUA都会得到完全的批准,即使得到了高层的支持。美国前总统唐纳德·特朗普(Donald Trump)对抗疟药羟氯喹的大力支持是导致其EUA在3月底通过的一个因素。但对SARS-CoV-2感染有效性证据不足,导致该EUA不到三个月后被撤销。这清楚地提醒,尽管监管结果可能结合了科学、患者需求和政治背景等因素,但最终还是证据会取得成功。
三、人血浆是抗感染药物开发计划的良好起点。自20世纪初以来,被动免疫被认为是治疗传染病的有效方法,SARS-CoV-2也证明了这一点。美国FDA为被动免疫疗法签发了三份EUA:恢复期患者血浆的收集和使用;礼来的单抗bamlanivimab;再生元的两种抗体混合物casirivimab和imdevimab。10月,美国FDA又批准了再生元的另一种被动免疫疗法Inmazeb,用于治疗埃博拉病毒的三种单抗的鸡尾酒疗法。
四、大多数小分子是对付高度传染性病毒性呼吸道病原体的“迟钝”的工具,但在缺乏替代品的情况下,也算足够了。吉利德Veklury(remdesivir)不是为SARS-CoV-2设计的,它是一种病毒RNA依赖性RNA聚合酶的抑制剂,具有广泛的体外抗病毒作用,可对抗多种病毒家族的动物和人类病原体。如果早期使用,它表现出了缩短SARS-Cov-2感染者的恢复时间。类似地,礼来Olumiant(baricitinib)是一种Janus激酶(JAK)小分子抑制剂,最初被批准用于治疗类风湿性关节炎,在COVID-19的第一波病毒中被发现具有抑制细胞因子风暴的作用,细胞因子风暴夺去了许多人的生命。
五、对EUAs来说,多数人的需要胜过少数人的需要,即创新服务于每个人。
美国FDA生物制剂评价与研究中心(CBER)主任彼得·马克斯(Peter Marks)说:“COVID-19提出了更好地将药企问题分类的需要。一些与COVID-19相关的文件明显表明,解决一个问题可以极大地帮助药企加快开发周期。在COVID-19之前,一家小公司将不得不等待75天——C型会议的标准时间表——才能获得一些或许并不那么重要的建议。这种等待的时间过长,今后我们希望找到增加互动的方法,这样药企的问题就不会恶化。”
另一个加快COVID-19产品评估的监管调整是,临床和生产方面完全并行地进行检查。展望未来,马克斯说:“特别是在疫苗和基因治疗领域,有可能通过先进的制造技术和非临床和临床项目的咨询过程显著缩短周期时间,特点是非正式的互动数量增加。”此外,2020年有明显证据表明,药物和生物制品的监管正在朝着两个新的理想方向发展:以患者为中心和监管的国际化。
监管全球化
目前已知的一个全球性监管倡议就是Orbis项目,由美国FDA领导,一些国家监管机构正在合作评估肿瘤领域的药物应用。在这个项目中,向美国FDA提交的一份监管文件将由各参与国家的专家集体评估,如果数据可以接受,将获得多国批准。但是,对批准的时间和精确性质的控制可能会有所不同。在联合评估之后,不同的国家机构可对其国家实施不同的标签和要求。实际上,Orbis项目是一条评估通道,它打开了多扇类似且几乎同时获得批准的大门,但它并没有强制执行统一的监管立场。
Orbis项目始于2019年9月,当时美国FDA与澳大利亚、加拿大监管部门批准了卫材(Eisai)Lenvima(lenvatinib)和默沙东Keytruda(pembrolizumab)联合治疗晚期子宫内膜癌。2020年,新加坡和瑞士的监管机构加入了Orbis项目,批准其首个新药Tukysa(tucatinib),一种由Seagen开发的HER2小分子抑制剂,与曲妥珠单抗和卡培他滨联合治疗不能切除或转移性HER2阳性乳腺癌。Tukysa从提交到批准仅用了119天,这是美国FDA实时肿瘤学审查程序下新分子实体的最快审批记录。
然而,细胞和基因治疗走向国际接轨却遭遇了阻碍。美国、欧洲和日本的监管机构于2020年1月在日内瓦举行了面对面会议,讨论这类疗法的联合审批需要延期做更多考量。马克斯说:“基因和细胞治疗项目的相对新颖性和稀有性可能意味着,不同国家监管的策略是不同的。但全球仍将向着相同的监管方法发展,只是目前经验尚不充足。我们将首先尝试调整一些技术要求,以便药企可以在任何地方使用相同的档案来支持相同的产品。”
以患者为中心
无论是对患者还是对医疗系统,方便是药品审批时看重的一个关键词。Inqovi就是一个例子;这种口服联合用药避免了注射地西他滨的使用。2020年最亮眼的产品要数诺华Leqvio(inclisiran),被欧洲批准为首个治疗低密度脂蛋白胆固醇(LDL-C)升高的siRNA药物,这可能标志着几十年来医药市场最根本的转变之一。因为到目前为止,siRNA药物都只作为罕见疾病的治疗方法一直备受关注,如Alnylam的Givlaari(givosiran)于2019年11月被批准用于急性肝卟啉症,Oxlumo(lumasiran)于2020年11月被批准用于原发性高草酸尿1型。但Leqvio把siRNA带入一个新的维度。
Leqvio针对前蛋白转化酶枯草杆菌素kexin 9(PCSK9)转录物,与抗PCSK9单抗在胆固醇降低市场上展开竞争,如赛诺菲的Praluent(alirocumab,一种人类IgG1单抗)和安进的Repatha(evolucumab,一种人类IgG2单抗)。后两款药物曾大张旗鼓地推出,但在市场上的表现令人失望。然而,即使Leqvio未能成为诺华希望和投资者需求的畅销药,2021年也将标志着siRNA作为一种技术与单抗一起实现从针对性强向制药主流的转变(图2)。
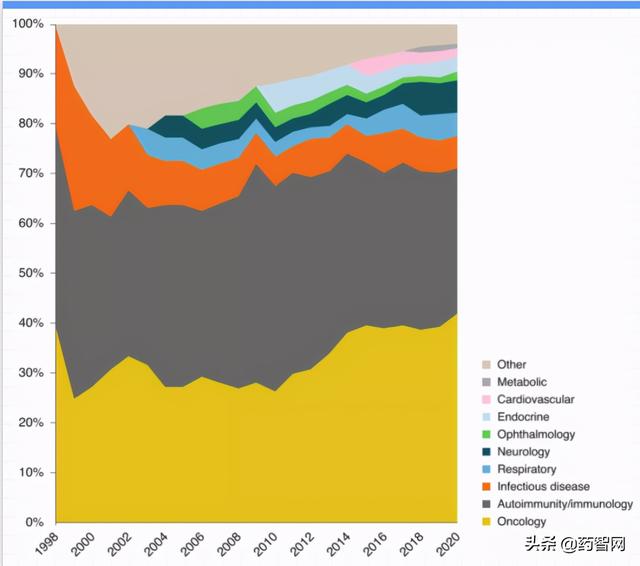
图2 不同疾病组的单克隆抗体随时间的批准情况
肿瘤学、免疫和自身免疫占主导地位,但其他疾病群体正在慢慢得到重视。
另一项凸显方便患者的新药审批是诺和诺德每周一次的自我注射人类生长激素(hGH)制剂Sogroya(somapacitan beco),这距离基因泰克的重组人生长激素Protropin(需要每天注射)获得批准过去了35年。诺和诺德使用了长效胰岛素类似物的技术关键:将药物循环半衰期从3-4小时延长到2-3天。
其他新药的批准将允许患者避免以医院为中心的治疗,这在COVID-19期间是一个特别理想的优势。如罗氏将来自Halozyme Therapeutics的重组人透明质酸酶添加到曲妥珠单抗和培妥珠单抗的固定剂量组合中,研发出Phesgo(培妥珠单抗/曲妥珠单抗/透明质酸酶-zzfx)。由于Phesgo是皮下注射,这意味着HER2阳性乳腺癌患者可以在输液中心外接受治疗。
COVID-19期间的安全考虑为罗氏的另一款产品Evrysdi(risdiplam)提供了快速打开市场的通道。该药于8月被批准用于治疗脊髓性肌萎缩症(SMA),是第三种批准用于SMA的药物,与渤健Spinraza和诺华Zolgensma不同,后两种药物分别通过鞘内和静脉给药,并且都需要住院治疗,Evrysdi是为婴儿配制的草莓味饮料,可以在家中服用。现在断言COVID-19和Evrysdi的推出如何改变SMA市场还为时过早,但值得注意的是,Spinraza在美国市场的销量已经在2020年第二季度下降了9%,第三季度比2019年同期下降了23%。
罕见病迎来重复批准
世界各地的监管政策优惠刺激了罕见病的药物开发。虽然孤儿药排他性可以为药物研发者提供经济激励,但低患者数量也带来了他们自己的难题,即招募患有严重遗传性疾病的患者进入安慰剂组的试验是否符合伦理或实践要求。美国FDA和其他监管机构表示同情,允许临床研究的对照组基于疾病进展的历史数据展开。用于Evrysdi批准的一项研究就使用了与未经治疗的SMA患者相比,证明该药物减缓独立坐姿或呼吸能力的丧失。
此外,极罕见病迎来首款新药。Hutchinson-Gilford早衰综合征处在罕见疾病谱的最末端,每1800万人中只有1人会受到影响。2020年11月,美国FDA批准了第一种治疗方法,来自Eiger BioPharmaceuticals的Zokinvy(lonafarnib),一种口服小分子法尼基转移酶抑制剂,平均延长了患者2.5年的寿命。
孤儿药监管政策的影响之一是,多种药物在短时间内被批准用于同一罕见适应症。如自身免疫性疾病视神经脊髓炎谱系障碍(NMOSD),在全球范围内每10万白人中约有1-2人患病,每10万黑人中约有5-10人。2019年至今已有三种疗法获批,都是单抗药物,分别为Alexion的Soliris(eculizumab),一种针对补体蛋白C5的人源化单克隆IgG2/4κ单克隆抗体;阿斯利康Uplizna(inebilizumab cdon),一种CD19导向的人源化无糖基化IgG1单抗;罗氏Enspryng(satralizumab-mwge),一种抗人白细胞介素-6受体的人源化IgG2单抗。这三种产品之间的竞争可能很激烈,因为在美国估计只有4000-8000人患有NMOSD。同时其他产品还在快速加入这一行列:Soliris下一代长效(每八周一次)制剂Ultomiris(ravulizumab cwvz),一种C5靶向IgG2/4κ单抗,含有一种设计用于降低新生儿Fc受体结合的Fc,正在进行3期研究(NCT04201262)。
美国瑞穗证券公司高级生物技术分析师杨迪飞(Difei Yang)表示:“在罕见疾病的基因治疗领域也存在产品竞争,这种产品一致性削弱了任何先到市场的优势,而且这在基因治疗的一次性世界中尤其重要。竞争最终会给病人和医生带来更好、更多的选择,但也可以预期,这会给新疗法的高价贴上一个保护标签。”
优化过继细胞疗法
2020年7月,美国FDA批准吉利德的Tecartus(brexucabtagene autoleucel),自体T细胞经逆转录病毒体外工程以表达包含与CD28和CD3ζ共刺激域相连的小鼠抗CD19单链可变片段(scFv)的CAR,用于治疗晚期套细胞淋巴瘤。这是吉利德2017年以119亿美元收购Kite Pharma后推出的第二款产品。与第一个批准的CAR-T疗法Yescarta(Axicabatgene ciloleucel)治疗大B细胞非霍奇金淋巴瘤相比,Tecartus的制造过程有额外的T细胞富集步骤,可减少自体细胞制剂中表达CD19的肿瘤细胞的扩增。实际上,Tecartus是一款“更纯净”的CAR-T产品。
尽管产品纯度更高,但给患者使用Tecartus仍然是一项繁重的工作,需要经过美国FDA认证的治疗中心,并配备经过培训的医务人员监管治疗的严重副作用。在目前的发展阶段,过继性细胞治疗仍然是癌症治疗的最后一步:Tecartus获得批准的试验中,所有患者以前不仅对化疗没有反应,而且对CD20抗体(如利妥昔单抗、奥克列珠单抗、奥比努珠单抗、veltuzumab或ofatumumab等单抗)或Bruton酪氨酸激酶抑制剂(小分子如伊布替尼、阿卡拉布替尼或扎努布替尼)治疗也没有反应。
皮肤下用药
大型制药公司对抗体产品的技术进步赞不绝口,因为它们不仅提高了药品的性能和方便性,而且为原研公司建立了知识产权保护。Halozyme的重组透明质酸酶平台Enhanze帮助生物药物的使用从专科中心(如医院或输液中心)重新定位到医生的办公室或家庭中。重组人透明质酸酶是一种具有通透性的酶,可分解皮肤和皮下组织细胞外基质中的皮下多糖,从而促进生物制剂的装载、分散和吸收,其允许通过皮下注射而不是输液给药。
两种装载Enhanze平台透明质酸酶的单克隆抗体产品于2020年获得批准:杨森Darzalex Faspro(daratumumab、靶向CD38的人IgG1κ单克隆抗体和透明质酸酶-fihj的混合物)用于治疗轻链淀粉样变病,以及罗氏Phesgo。
另外3种拥有Halozyme技术的批准药物为Baxalta的HyQvia(透明质酸酶和10%人多克隆免疫球蛋白输注);罗氏Rituxan Hycela(透明质酸酶和嵌合抗CD-20 IgG1κ单抗),和赫赛汀(曲妥珠单抗和透明质酸酶-oysk)。
抗体-药物结合物发展
另一个在2020年药物批准中表现突出的抗体辅助物是Seagen的抗体导向结合物(ADC)技术。4月,Immunomedics的Trodelvy(sacituzumab govitecan hziy,一种针对Trop-2的人源化IgG1κ单抗,通过可水解的CL2A连接物与拓扑异构酶抑制剂药物SN-38结合)批准用于三线治疗转移性三阴性乳腺癌。之后,葛兰素史克针对多发性骨髓瘤的Blenrep(belantamab mafodotin blmf,一种通过抗蛋白酶的马来酰亚胺丙丙烯酰连接物与微管抑制剂单甲基auristatin F共价连接的无氟酰化人源化IgG1单克隆抗体)批准。
Trodelvy被公司及其顾问称为“产品中的管线”,因为它代表了一种不同的ADC方法。早期的ADC(如Adcetris)将一个单一的、有毒的分子负载与抗体紧密结合。当结合抗体和毒素的复合物被内化并且连接蛋白降解时,肿瘤杀伤随之发生。相比之下,Trodelvy将一种中等毒性的化合物(SN-38)复制到一个抗体上。连接体的化学成分可以调节,这样,除了内化外,毒素在肿瘤的生理条件下局部释放。
Fc工程进入领先状态
7月底,Monjuvi(tafasitamab cxix,一种抗CD19人源化IgG1/2单抗,含有一个带有两个氨基酸替代物的混合Fc结构域,用于修饰Fc介导的功能)的加速批准使其成为MorphoSys的第二个获批产品,也是第二个批准使用Xencor的XmAb Fc域工程技术的治疗药物。对于Monjuvi,XmAb工程增加了vitro3中自然杀伤(NK)细胞介导的抗体定向细胞毒性(ADCC)的水平。Monjuvi的初始适应症仅限于一个狭窄的患者亚组——作为不符合自体干细胞移植条件的复发性或难治性弥漫性大B细胞淋巴瘤患者的二线治疗,必须与来那度胺(新基Revlimid)联合使用。然而,MorphoSys和美国合作伙伴Incyte希望将其应用扩展到一系列淋巴瘤适应症。
12月16日批准了第二种Fc工程化单克隆抗体:MacroGenics的Margenza(margetuximab cmkb),一种HER2靶向嵌合IgG1κ单抗,含有一种Fc工程抗体,用于增强活化Fcγ受体3A(CD16A)的结合,并降低与抑制性Fcγ受体2B(CD32B)的结合。这些变化导致体外ADCC和NK细胞的活化。Margenza在头对头试验中的表现优于曲妥珠单抗,尤其是在低亲和力CD16A基因型的患者中,曲妥珠单抗对这些患者的疗效较差。
基因靶向药物继续前进
所谓的精准抗癌药物是生物技术的一个交易热点,2020年批准了6款基因靶向产品(表3)。遗传标记的使用使临床开发成本得以降低,其方法是将开发计划针对少数经过高度筛选的患者。旗舰产品是诺华的格列卫(伊马替尼),这是一种小分子药物,2001年被批准用于治疗携带9号和22号染色体融合(费城染色体)的慢性粒细胞白血病(CML)。格列卫将5年生存率从30%提高到89%左右。
表3 2020年批准的基因靶向药物

2020年Blueprint的前两个产品获得批准,都是小分子多酪氨酸激酶抑制剂:Ayvakit(avapritinib),用于血小板源性生长因子受体第18外显子a D842V突变患者;Gavreto(普拉斯替尼),用于治疗RET基因融合(CCDC6-RET)和V804L、V804M和M918T突变的患者。这些批准中的大多数不会产生像格列卫那样的影响,因为特定的基因重排只占其他癌症的一小部分。例如对于甲基转移酶EZH2的小分子抑制剂Tazverik(tazemetostat)的开发者Epizyme来说,有限的患者群体(该药物仅被批准用于导致整合酶相互作用子1缺失的突变患者)意味着该公司可能难以偿还其在该药物上的投资。
2020年批准的另一种精准药物Retevmo(selpercatinib)是一种小分子多激酶抑制剂,用于治疗细胞外富含半胱氨酸区域(M918T、V804M、V804L等)RET突变的患者,其商业前景表现很好。Retevmo是礼来2019年初以80亿美元收购Loxo Oncology获得的重要资产。Gavreto则迎来了罗氏的投资,预付6.75亿美元,外加1亿美元的股权投资,用于非美国、非中国的共同开发和共同商业化权利。
瑞穗分析师杨说:“生物技术公司和大型制药公司之间的合作会持续下去。生物技术带来了技术创新的优势和临床验证的方法,使产品在最有价值的适应症或疾病环境中向前发展。另一方面,制药公司有足够的资金驾驭复杂的监管环境,投资于新的制造技术,并与付款人/医生/患者合作,使新疗法实现最佳商业化”(表4)。
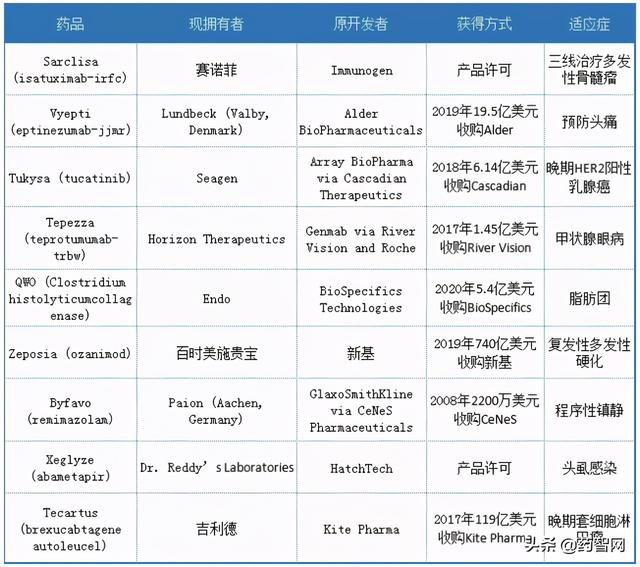
2020年批准药物名单中的授权药物
展望2021年
乔·拜登于2021年1月20日就任美国总统不太可能影响药品审批政策,但可能会影响定价。拜登支持医疗保险药品国际定价指数和旨在使药品价格与通胀同步的政策,这使得他在生命科学行业的药品定价改革方面可能比前总统更差。
目前,生物技术和制药行业的大部分公众注意力仍将集中在COVID-19相关疗法和疫苗的进展上。CBER马克斯认为,除了跟上生物创新的步伐外,产业界和监管机构应对大规模制造创新的挑战也是当务之急。对于抗体,甚至是疫苗,我们能看到连续或半连续的生产而不是批量生产,我们需要为此努力,以便为下一次流行做好更好的准备。”。
在投资者和行业内,计划于2021年第一季度作出的关键监管决定将有助于明确人源化单克隆抗体在实质性适应症(如阿尔茨海默病、骨关节炎和代谢性疾病)中的作用(图2)。
此外,越来越多的降胆固醇药物的治疗选择,包括Leqvio的推出,预示着一个竞争日益激烈的市场。再生元的抗血管生成素样3蛋白的全人类IgG1单抗evinacumab审批申请预计将于2021年2月提交美国FDA,并将成为首个用于纯合家族性高胆固醇血症的单抗。
3月,美国FDA将审查辉瑞和礼来的人源化IgG2重链和κ-轻链单抗tanezumab,这是针对神经生长因子(NGF)的后期抗体药物(还有一种是再生元和Teva的全人类IgG1单抗fasinumab)。抗NGF药物的主要问题是,导致患者的骨关节炎病情恶化。这一研究结果限制了两者单抗药物的临床方案只能使用低剂量。由于病情恶化,tanezumab治疗膝关节和髋关节骨关节炎的两项早期试验分别于2009年和2010年停止,最终该药的任何批准可能仅限于不能使用标准护理止痛药(如非甾体抗炎药)的患者。
6月,美国FDA预计将最终宣布对渤健(Biogen)阿尔茨海默病药物aducanumab(一种针对β-淀粉样蛋白的嵌合人IgG1单抗)的决定。Stifel分析师Matteis称:“这一决定关系重大。除了aducanumab可能代表第一种治疗阿尔茨海默病的药物外,它的成败还将对渤健的盈利能力产生深远影响。鉴于咨询委员会的否定意见,美国FDA批准这种药物将是‘相当有争议的’。但投资者可能会认为FDA更为通融,向阿尔茨海默症患者的其他公司发出积极的信号,因为大量的医疗需求仍得不到满足。”
参考文献:
1. Refreshing the biologic pipeline 2020
2. Selumetinib in Children with Inoperable Plexiform Neurofibromas
3. The Fc-engineered CD19 antibody MOR208 (XmAb5574) induces natural killer cell-mediated lysis of acute lymphoblastic leukemia cells from pediatric and adult patients
4. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC)
![]()
责任编辑:三七
声明:本文观点仅代表作者本人,不代表药智网立场,欢迎在留言区交流补充;如需转载,请务必注明文章作者和来源。